Болезнь Фабри
| Болезнь Фабри | |
|---|---|
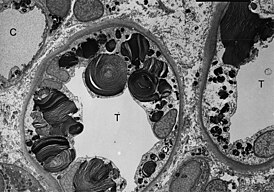 ТЭМ-микроскопия показывает наличие включений гликосфинголипидов различной формы и размеров в клетках дистальных канальцев почки. | |
| МКБ-10 | E75.2 (ILDS E75.25) |
| МКБ-10-КМ | E75.21 и E75.2 |
| МКБ-9 | 272.7 |
| OMIM | 301500 |
| DiseasesDB | 4638 |
| eMedicine | neuro/579 derm/707 ped/2888 |
| MeSH | D000795 |
Боле́знь Фабри́ (болезнь Андерсона—Фабри, англ. Anderson—Fabry disease) — редкое генетически детерминированное заболевание с Х-сцепленным типом наследования, из группы лизосомных болезней накопления. Ранее считалось, что тип наследования болезни Фабри — X-сцепленный рецессивный, однако на современном этапе накоплено достаточно данных, чтобы считать тип наследования болезни Фабри X-сцепленным доминантным с неполной пенетрантностью у женщин. Данное заболевание вызвано нарушением метаболизма сфинголипидов и обладает широким спектром клинических симптомов.
Содержание
- 1 Историческая справка
- 2 Эпидемиология
- 3 Наследование
-
4 Клиническая картина
- 4.1 Внешний вид больных
- 4.2 Боль
- 4.3 Нарушения потоотделения
- 4.4 Быстрая утомляемость и непереносимость физических нагрузок
- 4.5 Ангиокератомы
- 4.6 Воронковидная (вихревая) кератопатия
- 4.7 Нарушения функции сердца
- 4.8 Нарушение функции почек
- 4.9 Мозг и нервная система
- 4.10 Слух
- 4.11 Проблемы в психоэмоциональной сфере
- 5 Диагностика
- 6 Лечение
- 7 Прогноз
- 8 См. также
- 9 Примечания
- 10 Ссылки
Историческая справка
Эпоним
Болезнь названа в честь одного из его первооткрывателей — Джона Фабри (1 июня 1860 г. — 29 июня 1930 г.).
Впервые заболевание было описано независимо двумя дерматологами Джоном Фабри (1860—1930) из Германии и Вильямом Андерсоном (1842—1900) из Великобритании:
- в 1898 году Фабри описал 13-летнего мальчика с нодулярной пурпурой, у которого впоследствии развилась альбуминурия. Данный клинический случай был классифицирован автором как один из вариантов диффузной ангиокератомы.
- В том же году Андерсон описал 39-летнего мужчину с ангиокератомой, протеинурией, деформациями пальцев рук, варикозным расширением вен и лимфатическим отёком.
Эпидемиология
Распространённость данного заболевания составляет от 1 на 40 000 до 1 на 120 000 живых новорождённых. Таким образом, является одной из наиболее распространённых (после болезни Гоше) лизосомных болезней накопления, встречается во всех расовых группах и возникает с частотой 1 на 117 000 в Австралии, 1 на 476 000 в Нидерландах, 1 на 40 000—60 000 мужчин в США. Более распространено лёгкое, атипичное течение заболевания с наличием признаков поражения одного органа. Обычно (примерно в 95% случаев) больные наследуют дефектный ген от одного из родителей (мужчина - от матери, женщина - от матери или отца), но около 5% случаев связаны с так называемыми мутациями de novo. Таким образом, отсутствие семейной истории заболевания не исключает наличия болезни Фабри.
Наследование
Клиническая картина болезни Фабри обусловлена мутациями в гене GLA, расположенном на длинном плече Хq22 и кодирующим фермент α-галактозидазу А. Механизм наследования — сцепленный с полом:
- Мужчины (обладатели одной дефектной Х-хромосомы) с классическим фенотипом болезни Фабри по законам генетики способны передать мутантную хромосому только своим дочерям.
- Женщины гетерозиготны (имеют одну нормальную и одну дефектную хромосому). По законам Менделя они передают мутантный ген половине своих потомков. При этом, течение болезни у них отличается умеренно-выраженным характером с более поздним началом, медленным прогрессированием и наличием лёгких клинико-патологических изменений. Тем не менее, рядом исследований продемонстрировано наличие признаков тяжёлого течения болезни и у гетерозиготных женщин. При этом механизм, посредством которого у гетерозиготных женщин развиваются клинические симптомы болезни Фабри, до конца не известен. Наиболее вероятной причиной является неслучайная лайонизация. У большинства таких женщин определяется практически нормальный уровень циркулирующего в крови фермента, а случайный процесс инактивации одной Х-хромосомы подразумевает мозаичность их тканей, которые состоят как из нормальных, так и ферменто-дефицитных клеток.
Клиническая картина
Постоянно пополняющаяся Международная база данных по болезни Фабри содержит информацию примерно о 1200 пациентах. Симптомы болезни нередко проявляются в раннем детстве, однако, ввиду своей неспецифичности, задержка с диагнозом зачастую составляет годы и десятилетия. С возрастом количество симптомов и степень их выраженности обычно увеличивается.
Различают классическую и неклассическую форму болезни Фабри.
Внешний вид больных
Мужчины зачастую обладают характерным фенотипом, напоминающим внешний вид пациентов с акромегалией: выступающие супраорбитальные дуги и лобные бугры, выступающая нижняя челюсть, увеличенные губы, запавшая переносица (с 12—14-летнего возраста).
Боль
Боли необычного характера - один из самых частых и ранних симптомов болезни Фабри.
Выделяют два основных типа болей:
- Нейропатическая боль (акропарестезии). Постоянное жжение и покалывание, ощущение дискомфорта. Эти боли обычно затрагивают ступни и ладони.
- Кризы Фабри. Возникающие время от времени сильные, жгучие боли, возникающие в ступнях или ладонях и иногда распространяющиеся на другие части тела. Эти боли могут быть весьма изнуряющими и продолжаться от нескольких минут до нескольких дней.
Изменения погоды, перегрев тела, стресс, физические упражнения и усталость могут провоцировать усиление боли.
Нарушения потоотделения
Значительная часть больных потеет меньше нормы (гипогидроз) или не потеет вообще (ангидроз). Это может приводить к перегреву тела и непереносимости жары. Предполагается, что причина этого нарушения кроется в повреждении нервов и клеток потовых желез.
Быстрая утомляемость и непереносимость физических нагрузок
Некоторые больные не переносят физических нагрузок, быстро устают, испытывают перегрев даже при умеренных нагрузках. К тому же, физические нагрузки могут провоцировать приступы боли.
Ангиокератомы
Один из самых ранних и заметных признаков болезни Фабри - мелкие, красновато-фиолетовые безболезненные папулы на коже, называемые "ангиокератомы". С возрастом количество ангиокератом у больных увеличивается, также иногда увеличиваются размеры отдельных элементов сыпи (до 10 мм). Наиболее часто ангиокератомы располагаются в области губ, пальцев рук и ног, ано-генитальной области (при достижении половой зрелости). Важно помнить, что наличие ангиокератом - не патогномоничный признак болезни Фабри, а также наблюдается при врожденной геморрагический телеангиэктазии (болезнь Рандю-Вебера-Ослера), болезни Фордайса, болезни Шиндлера (наследственный дефицит лизосомального фермента N–ацетил–альфа–D–галактозаминидазы), фукозидозе и сиалозидозе.
Воронковидная (вихревая) кератопатия
При офтальмологическом осмотре с применением щелевой лампы у больных Фабри нередко выявляются тусклые золотистые вихреобразные отложения субстрата в роговице глаза. На остроту зрения эти отложения, как правило, не влияют. При дифференциальном диагнозе нужно учитывать, что длительное применение амиодарона вызывает такие же изменения в роговице, исчезающие с прекращением приема препарата.
Нарушения функции сердца
Накопление субстрата в клетках сердечно-сосудистой системы приводит к различным нарушениям, являющихся одной из основных причин смерти при болезни Фабри. Больные жалуются на затруднение дыхания и боли в груди, боли по типу стенокардитических, учащенное сердцебиение. Наиболее часто наблюдается гипертрофия левого желудочка, аритмии.
Нарушение функции почек
Нарушение функции почек наблюдается у значительной части детей, многих женщин и большинства мужчин с болезнью Фабри и в конечном итоге приводят к терминальной стадии хронической почечной недостаточности у большинства больных мужчин и некоторых женщин. Наряду с сердечно-сосудистыми нарушениями почечная недостаточность является одной из основных причин смерти при болезни Фабри. Начальные стадии почечной недостаточности остаются практически незамеченными (больной не предъявляет жалоб). В отличие от "классических" нефрологических больных, у больных Фабри обычно нормальное артериальное давление, нормальный или около нормального уровень сывороточного креатинина, "минимальная" протеинурия, что затрудняет оценку степени почечной недостаточности нефрологом, недостаточно опытным в области диагностики и лечения болезни Фабри. Для мониторинга и оценки функции почек всем больным Фабри необходимо по меньшей мере ежегодно проходить серию тестов, включающих измерение белка в моче, креатинина в сыворотке и моче, скорость клубочковой фильтрации и анализ суточной мочи.
Мозг и нервная система
Болезнь Фабри имеет много неврологических проявлений. Больные жалуются на головокружение, головные боли. Нарушение мозгового кровообращения вследствие накопления сфинголипидов выливается в транзиторные ишемические атаки, инсульты. Иногда ранний инсульт является первым и единственным симптомом болезни Фабри, что необходимо учитывать при расследовании причин всех ранних инсультов у женщин и мужчин. Ишемия сосудов головного мозга - основной механизм поражения центральной нервной системы при болезни Фабри; характерны ишемические инсульты, преимущественно в вертебробазиллярном бассейне.
Слух
Болезнь может влиять на слух. Пациенты могут жаловаться на звон в ушах, а в отдельных случаях наблюдается ухудшение слуха, которое может медленно прогрессировать или появиться внезапно.
Проблемы в психоэмоциональной сфере
Жизнь с тяжелым болевым синдромом зачастую негативно сказывается на душевном состоянии больных. Нередки депрессии, тревожные расстройства. По результатам исследования 2014 года, пациенты мужского пола демонстрируют снижение скорости обработки информации и способностей к целенаправленной деятельности.
Диагностика
Диагностика болезни Фабри включает комплексную оценку клинический картины, лабораторные тесты и другие исследования (например, исследование биоптата почки на наличие подоцитов, лизосомы которых заполнены специфическим субстратом, характерным для болезни Фабри).
На МРТ головного мозга могут присутствовать характерные для болезни Фабри признаки: на Т2-изображении может присутствовать гиперинтенсивный сигнал в белом веществе фронтальных и теменных долей. На Т1-взвешенном изображении наблюдается высокий сигнал от серого вещества глубинных структур, особенно заднего бугорка таламуса. Изолированное поражение заднего бугорка таламуса считается патогномоничным для болезни Фабри. Также на МР-картине часты сосудистые мальформации, преимущественно представленные долихоэктазиями вертебробазилярной артерии.
На ЭКГ наблюдается гипертрофия левого желудочка, укорочение P-R интервала (на ранних стадиях болезни) и предсердножелудочковая блокада (на более поздних стадиях). На Эхо-КГ: преимущественно концентрическая гипертрофия левого желудочка, а в некоторых случаях - гипертрофия правого желудочка и папиллярной мышцы. На МРТ сердца наблюдается позднее поступление контраста (гадолиний) во внутреннюю стенку левого желудочка.
Разрабатываются специальные опросники, которые с высокой чувствительностью и специфичностью позволяют врачам выявлять пациентов с высокой вероятностью наличия болезни Фабри.
Для лабораторной диагностики болезни Фабри применяются следующие исследования:
Определение активности альфа-галактозидазы (энзимодиагностика)
При болезни Фабри активность альфа-галактозидазы в крови у мужчин всегда снижена, а у женщин активность GLA может быть около нижней границе нормы, чуть ниже её или нормальной, из-за чего ферментный анализ для женщин не показателен в отношении наличия/отсутствия болезни Фабри. В качестве материала для исследования используется как жидкая кровь (в пробирке с гепарином или ЭДТА), так и сухие пятна крови. Последние очень удобны для скрининговых исследований (например, неонатального скрининга), так как сухие пятна крови легко получать, хранить и транспортировать. Активность фермента определяют флуориметрическим методом или с помощью MS-MS.
Секвенирование экзонов и приэкзонных участков интронов гена GLA (ДНК-диагностика)
Идентифицировано более четырёхсот мутаций, приводящих к развитию болезни Фабри, большинство из которых являются уникальным для каждой семьи. У больного мужчины мутация выявляется в гемизиготном состоянии, у женщины - в гетерозиготном состоянии. Секвенирование ДНК гена GLA - наиболее точный метод диагностики болезни Фабри, но он не применим для широкомасштабного скрининга, не может быть использован в качестве первичного теста ввиду высокой стоимости (примерно в 5 раз дороже энзимодиагностики). Для женщин предпочтительно проведение ДНК-диагностики, т.к. энзимодиагностика не всегда позволяет выявить болезнь. При подтверждении диагноза и выявлении мутации у пробанда целесообразно обследовать на наличие этой мутации всех родственников пробанда, которые могут нести одну с ним X-хромосому.
Количественное определение сфинголипидов
Относительно новый тест, рекомендованный для использования в комплексной лабораторной диагностике болезни Фабри. Количественное определение глоботриазилсфингозина (Lyso-GB3, Lyso-GL3) более показательно, нежели глоботриазилцерамида (GB3, GL3). Тест позволяет не только разъяснить трудные диагностические случаи болезни, но и определить наиболее вероятную форму болезни (классическая или неклассическая). Полезен для мониторинга состояния пациента, т.к. показано, что улучшение на фоне патогенетической терапии сопровождается снижением содержания Lyso-GL3 в плазме пациентов. В качестве материала для исследования можно использовать не только плазму крови, но и более удобные сухие пятна крови. Определение Lyso-GL3 в сухом пятне крови выполняет лаборатория ARCHIMEDlife (Вена, Австрия).
Диагностика болезни Фабри в России
В России ферментную и генетическую диагностику болезни Фабри выполняют в лаборатории наследственных болезней обмена веществ МГНЦ РАМН и в лаборатории молекулярно-генетической диагностики Научного центра здоровья детей.
Лечение
При болезни Фабри применяют симптоматическую терапию (например, для купирования болей могут применяться анальгетики, антиконвульсанты, НСПВП) и этиотропную ферментозаместительную терапию (ФЗТ), которая направлена на снижение выраженности и предотвращение симптомов болезни Фабри. В России зарегистрированы два лекарственных препарата для ФЗТ болезни Фабри: агалсидаза-бета (Фабразим®, Джензайм) и агалсидаза-альфа (Реплагал®, Шайер).
Прогноз
При своевременном доступе к ФЗТ, надлежащем мониторинге болезни и соблюдении врачебных рекомендаций прогноз благоприятный.
Женщины с болезнью Фабри могут иметь детей. Недавнее исследование показало, что некоторые симптомы болезни Фабри могут осложнить течение беременности и послеродовой период (болезненные симптомы со стороны ЖКТ, акропарестезии, протеинурия, головные боли и послеродовая депрессия). Хотя жизнеугрожающих осложнений беременности и родов не было выявлено, было показано статистически значимое увеличение частоты развития гипертензии среди беременных женщин с болезнью Фабри.