Лизосомные болезни накопления
| Лизосомные болезни накопления | |
|---|---|
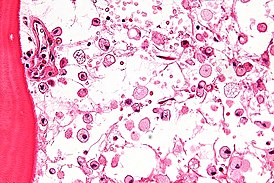 На микрофотографии характерные изменения строения костного мозга при болезни Гоше — цитоплазма макрофагов напоминает смятую папиросную бумагу. Окраска гематоксилином и эозином | |
| МКБ-10 | E75.-E77. |
| MeSH | D016464 |
Лизосо́мные боле́зни накопле́ния (англ. Lysosomal Storage Diseases) — общее название группы весьма редких наследственных заболеваний, вызванных нарушением функции внутриклеточных органелл лизосом. Эти одномембранные органоиды являются частью эндомембранной системы клетки и специализируются на внутриклеточном расщеплении веществ: гликогена, гликозаминогликанов, гликопротеинов и других. Лизосомные болезни накопления вызываются генетически обусловленным дефицитом ферментов лизосом, что приводит к накоплению макромолекул, являющихся субстратом этих ферментов, в различных органах и тканях организма.
Данная группа объединяет мукополисахаридозы, муколипидозы, гликогенозы, болезни накопления липидов, гликопротеинов и других макромолекул.
Содержание
Историческая справка
Клиническая картина первого наследственного заболевания из группы лизосомных болезней накопления (болезнь Тея — Сакса) была описана в 1881 году.
Затем, в 1882 году описано заболевание, названное в честь впервые описавшего его французского врача Филиппа Гоше.
В 1932 году голландский врач Иоанн Помпе описал гликогеноз второго типа, впоследствии названный по его имени болезнью Помпе.
В конце 1950-х — начале 1960-х годов бельгийский биохимик Кристиан де Дюв с соавторами, используя методику фракционирования клеток, открыл лизосомы в качестве клеточных органелл, ответственных за расщепление и утилизацию макромолекул. Данное научное открытие дало возможность вскоре выявить патофизиологическую основу лизосомных болезней накопления.
Болезнь Помпе стала первым наследственным заболеванием, идентифицированным как лизосомная болезнь накопления. В 1963 году бельгийский физиолог и биохимик Генри Хэрс (англ. Henri G. Hers) опубликовал работу, в которой связал причину развития данного симптомокомплекса с дефицитом α-глюкозидазы и высказал предположение о связи других генетических заболеваний, в том числе мукополисахаридозов, с недостаточностью того или иного фермента.
Эпидемиология
По состоянию на 2014 год, известно свыше 50 лизосомных болезней накопления, среди 7000—8000 новорождённых диагностируют около одного заболевания этой группы. Каждое заболевание индивидуально встречается не чаще, чем у одного из 100 тысяч новорождённых, при этом распространённость конкретного заболевания в различных популяциях может значительно варьировать. Распространение мутаций, ведущих к какому-либо заболеванию из группы лизосомных болезней накопления, определяется популяционно-генетическими факторами и, в первую очередь, связано с эффектом основателя. Типичным следствием эффекта основателя являются различие по наиболее распространённым мутациям одного и того же гена в различных генетически изолированных популяциях и этнических группах.
Наследование
Подавляющее большинство лизосомных болезней накопления наследуются аутосомно-рецессивно, за исключением трёх заболеваний, которые наследуются сцепленно с полом. К этим трём заболеваниям относятся мукополисахаридоз Хантера (МПС II) и болезнь Фабри, являющиеся рецессивными Х-сцепленными заболеваниями, а также синдром Данона, являющийся доминантным Х-сцепленным заболеванием.
Патогенез
Большая часть заболеваний группы связана с генетически обусловленным нарушением функции какой-либо из лизосомных гидролаз. Это приводит к прогрессивному накоплению внутри клетки промежуточного субстрата, который в норме распадается. Некоторые болезни этой группы вызваны нарушениями белков, участвующих в везикулярном транспорте или в биогенезе лизосом. Например, муколипидоз II типа (I-клеточная болезнь) вызван дефектом фосфоэстеразы, которая локализована в аппарате Гольджи, нарушение работы этого фермента приводит к ошибочной переадресации лизосомных гидролаз во внеклеточное пространство вместо их транспорта из аппарата Гольджи в лизосому. Хотя ферменты лизосом относятся к белкам, которые экспрессируются в подавляющем большинстве типов клеток, аномальное накопление субстратных макромолекул происходит в тех клетках, тканях и органах, которые характеризуются повышенной скоростью обновления этих макромолекул. Первичное накопление макромолекул может вести к вторичным нарушениям других биохимических и клеточных функций, что обуславливает обычно тяжёлое течение заболеваний этой группы.
Классификация
В связи с тем, что при многих лизосомных болезнях накопления наблюдается сходная клиническая картина, их классифицируют в соответствии с типом вещества, которое накапливается (например, мукополисахаридозы, гликопротеинозы, сфинголипидозы).
Ниже приведена классификация, согласно действующей МКБ (указан код МКБ-10):
- E72. Другие нарушения обмена аминокислот.
-
E74. Другие нарушения обмена углеводов.
- E74.0 Болезни накопления гликогена. Болезнь Помпе.
-
E75. Нарушения обмена сфинголипидов и другие болезни накопления липидов.
- E75.0 GM2 ганглиозидоз: Болезнь Сандхоффа, Болезнь Тея — Сакса, GM2-ганглиозидоз: БДУ (без дополнительных уточнений), взрослых, ювенильный.
- E75.1 Другие ганглиозидозы. Ганглиозидоз GM1, Муколипидоз IV.
- E75.2 Другие сфинголипидозы. Болезнь Фабри. Болезнь Гоше. Болезнь Краббе. Болезнь Ниманна — Пика. Синдром Фарбера. Метахроматическая лейкодистрофия. Множественная сульфатазная недостаточность.
- E75.4 Липофусциноз нейронов. Болезнь: Баттена, Бильшовского — Янского, Куфса, Шпильмейера — Фогта
- E75.5 Другие нарушения накопления липидов. Болезнь Вольмана.
-
E76. Нарушения обмена гликозаминогликанов
- E76.0 Мукополисахаридоз, тип I. Синдромы: «Гурлер», «Гурлер — Шейе», «Шейе».
- E76.1 Мукополисахаридоз, тип II Синдром Хантера
- E76.2 Другие мукополисахаридозы. Недостаточность β-глюкуронидазы. Мукополисахаридозы типов III, IV, VI, VII. Синдром: Марото — Лами (лёгкий), (тяжёлый), Моркио(-подобный), (классический), Санфилиппо (тип A) (тип В) (тип С) (тип D)
-
E77. Нарушения обмена гликопротеинов.
- E77.0 Дефекты посттрансляционной модификации лизосомных ферментов. Муколипидоз II (I-клеточная болезнь), Муколипидоз III (псевдополидистрофия Гурлер).
- E77.1 Дефекты деградации гликопротеидов. Аспартилглюкозаминурия. Фукозидоз. Маннозидоз. Сиалидоз (муколипидоз I)
Диагностика
Разработаны специальные методы диагностики, опирающиеся на ряд постоянных признаков, характеризующих болезни накопления липидов:
- накопление в тканях сложных липидов, структурным компонентом которых является церамид;
- скорость синтеза запасаемого липида сравнима со скоростью его биосинтеза у здоровых людей;
- на фоне заболевания наблюдается недостаток специфичного фермента в лизосомах, необходимого для гидролиза липида;
- степень снижения активности фермента во всех тканях одинакова.
Отныне стало возможным выявление в популяции гетерозиготных носителей дефектных генов, ответственных за развитие данных заболеваний, а также выявлять сфинголиподистрофию у плода.
Клиническая картина
Предпосылками для проявления лизосомных болезней накопления являются различные генетические дефекты, которые ведут к развитию ферментопатии — недостаточности определённых ферментов, расщепляющих некоторые макромолекулы на уровне внутриклеточных органелл (лизосом). Лизосомные болезни накопления характеризуются:
- прогрессирующим течением,
- высокой инвалидизацией,
- высокой смертностью пациентов.
Наиболее характерными общими особенностями клинической картины для большинства лизосомных болезней накопления являются:
- органомегалия (преимущественно гепатомегалия и спленомегалия),
- костные аномалии,
- различной степени выраженности нарушения со стороны центральной нервной системы,
- грубые особенности строения волос и лица.
Лечение
До недавнего времени терапия наследственных болезней накопления носила исключительно паллиативный характер. Развитие науки позволило с 90-х годов XX столетия приступить к клинической коррекции лизосомных болезней накопления методом эффективной и безопасной ферментозаместительной терапии (англ. Enzyme Replacement Therapy). Принцип данного метода терапии сводится к введению в организм пациента модифицированной формы скомпрометированного генетической патологией фермента, обладающего нормальной активностью. Модификация формы дефектного фермента способствует повышенной проницаемости его в клетки тканей-мишеней, где непосредственно осуществляется процесс катализа реакции гидролиза субстратов накопления. Однако, в связи с коротким (несколько десятков часов) периодом полужизни фермента в клетке, ферментозаместительную терапию необходимо проводить на протяжении всей жизни пациента.