Синдром Элерса — Данлоса
| Ehlers-Danlos Syndrome | |
|---|---|
 | |
| МКБ-10 | Q79.6 |
| МКБ-10-КМ | Q79.6 |
| МКБ-9 | 756.83 |
| МКБ-9-КМ | 756.83 |
| OMIM | 130000 |
| DiseasesDB | 4131 |
| MedlinePlus | 001468 |
| eMedicine | derm/696 ped/654 |
| MeSH | D004535 |
Синдром Э́лерса — Данло́са (синдром Э́лерса — Данло́, син. Э-Д; англ. Ehlers-Danlos Syndrome, «гиперэластичность кожи» («Cutis hyperelastica»), несовершенный десмогенез, несовершенный десмогенез Русакова, синдром Черногубова — Элерса — Данлоса) — это группа наследственных системных заболеваний соединительной ткани, вызванных дефектом в синтезе коллагена. В зависимости от отдельной мутации, серьёзность синдрома может измениться от умеренного до опасного для жизни. Лечения нет, но существует терапия (уход), смягчающая последствия.
Синдром назван в честь двух дерматологов, идентифицировавших его в начале XX века: Эдварда Элерса (1863—1937) из Дании и Анри-Александра Данлоса (1844—1912) из Франции.
Содержание
Симптоматика

|

|
|
|
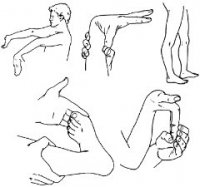
Человек с син. Э-Д демонстрирует гиперподвижность суставов.
| ||
Симптомы сильно варьируются в зависимости от типа болезни. Тем не менее, в каждом случае они вызваны повреждением или недостатком коллагена типа III. Болезнь как правило поражает суставы, кожу и кровеносные сосуды, с симптомами такими как свободные (плохо прикреплённые), сильно гнущиеся суставы; гладкая или эластичная, легко повреждающаяся кожа; неправильное заживление ран и формирование шрамов; маленькие и хрупкие кровеносные сосуды. Все формы затрагивают суставы, вызывая гиперподвижность, они выходят за пределы нормального диапазона движений. В результате люди с «син. Э-Д» более подвержены различным травмам таким как: вывихи, подвывихи, растяжения связок, деформация и иногда разрыв мягких тканей. Так как синдром часто не диагностируется, некоторые случаи принимаются за жестокое обращение с детьми.
Классификация
В прошлом было более десяти общепризнанных типов «син. Э-Д». В 1997 исследователи предложили более простую классификацию, которая сократила число главных типов до шести и дала им описательные имена. Другие типы условно могут существовать, но о них сообщается только в отдельных семьях или они плохо характеризованы. За исключением типа гиперподвижности, были идентифицированы и отождествлены отдельные мутации — они могут быть точно определены генетическим анализом. Это особенно важно из-за значительной вариативности признаков в индивидуальном рассмотрении, из-за чего может быть перепутана классификация только на симптоматической основе.
По распространенности среди населения:
| Имя | Номер | Описание | OMIM | Ген(ы) |
| Гиперподвижность (Hypermobility) | тип 3 | Поражает 1 человека на 10 000 - 15 000, вызван аутосомным доминирующим механизмом. Возникает в результате мутации любого из двух генов, которые вызывают Сосудистый тип и син. Э-Д с дефицитом тенасцина-X, соответственно. Проявляется в гиперподвижности суставов; поражение кожи выражено меньше. Характерны хронические мышечно-скелетные боли. | 130020 | COL3A1, TNXB |
| Классический (Classical) | тип 1 и 2 | Поражает приблизительно от 2 до 5 человек на 100 000. Затрагивает коллаген типа V, также коллаген типа I. | 130000, 130010 | COL5A1, COL5A2, COL1A1 |
| Сосудистый (Vascular) | тип 4 | Поражает приблизительно 1 человека на 100 000. Вызван аутосомно-доминантным дефектом в синтезе коллагена типа III | 130050 | COL3A1 |
| Кифосколиоз (Kyphoscoliosis) | тип 6 | Аутосомно-доминантный дефект, вызывающий недостаток фермента, называемого Лизин Гидролаза[1]. Очень редок; описано немногим более 60 случаев. | 225400, 229200 | PLOD1 |
| Артрохалазия (Arthrochalasis) | типы 7A и B | Поражает коллаген типа I. Крайне редок, описано всего около 30 случаев. | 130060 | COL1A1, COL1A2 |
| Дерматоспараксис (Dermatosparaxis) | тип 7C | Также крайне редок, описано около 10 случаев. | 225410 | ADAMTS2 |
Тип Гиперподвижность
Прежний тип 3, встречается у 1 человека на 10 000 — 15 000, что делает его самым распространённым вариантом болезни. Признаки и симптомы могут быть не диагностированы (не признаны) врачами или, как правило, ошибочно диагностированы как фибромиалгия и обычно больным не ставят диагноз, пока не проявятся серьёзные осложнения. Диагностика осуществляется, в основном, на клинических наблюдениях. Основные признаки и симптомы включают в себя:
- Свободные, нестабильные суставы, подверженные: растяжениям, вывихам, подвывихам (частичный вывих), переразгибание суставов
- Плоскостопие
- Высокое и узкое нёбо
- Лёгкие кровоподтёки
- Легко повреждающаяся бархатно-гладкая кожа
- Раннее начало остеопороза (обычно проявляется в середине 30 лет)
- Поражение сердца: в некоторой степени en:Dysautonomia или приобретённый порок сердца (такой как пролапс митрального клапана, что создаёт повышенный риск инфекции (эндокардит) во время операций (также возможно развитие до крайне опасной для жизни степени при пролапсе митрального клапана).
Другие симптомы и осложнения могут включать в себя:
- Низкая плотность костей (остеопения) — предшественник остеопороза
- Мышечная слабость, часто усугубляется холодной погодой
- Деформации позвоночника, такие как: сколиоз (искривление позвоночника), кифоз (горб в грудном отделе), en:Tethered spinal cord syndrome, en:Basilar invagination (cranial settling), а также Мальформация Арнольда — Киари (поражение продолговатого мозга, мозжечка выраженные затылочными болями, нарушениями глотания, атаксией и другими симптомами)
- Функциональные расстройства кишечника (функциональный гастрит, синдром раздражённого кишечника)
- Сдавление нервов (синдром запястного канала, парестезия, невралгия тройничного нерва)
- Болезнь Рейно
- Миалгия (боль в мышцах) и артралгия
- Чрезмерная усталость
- Преждевременный разрыв амниона (выкидыш) во время беременности en:Premature rupture of membranes
- Младенцы с гипервподвижностью суставов имеют слабый мышечный тонус (мышечная гипотония), который может задержать развитие таких моторных навыков как самостоятельное присаживание, вставание и хождение.
Боль, сопутствующая этому состоянию, является серьёзным осложнением.
Классический тип
При старой системе классификации он был разделён на два типа: тип I (тяжёлый) и тип II (умеренный). Поражает приблизительно от 2 до 5 человек на 100 000, и является вторым по распространённости. Поражает коллаген V и I типа. Важные симптомы затрагивают кожу и суставы. Больные как правило проявляют:
- гладкая, сильно эластичная, легко ранимая кожа
- уродливые или необычайно обширные шрамы, особенно на лбу, коленях, локтях и подбородке
- гиперподвижные суставы имеют тенденцию к вывихам, растяжению связок и подвывихам (обычно в коленной чашечке, в плече, в пястно-фаланговом суставе en:Metacarpophalangeal joint, и в височно-челюстном суставе
- Из-за сниженного мышечного тонуса, у младенцев может быть нарушено развитие моторных навыков
- Дети могут иметь склонность к развитию грыжи или смешению любого внутреннего органа.
В настоящее время не существует определённого теста для диагностики этого типа синдрома. И ДНК анализ и биохимические исследования используются для выявления пораженных болезнью. В некоторых случаях биопсия кожи была признана полезной при постановке диагноза. К сожалению эти тесты не достаточно надёжны, чтобы выявить всех больных. Если в семье есть несколько больных членов, то можно провести внутриутробную ДНК диагностику.
Сосудистый тип
Поражает приблизительно 1 человека на 100 000, вызван аутосомным доминантным дефектом в синтезе коллагена типа III. В старой системе классификации имел номер IV, этот тип является самой опасной разновидностью синдрома. Проведенные исследования определяют ожидаемую продолжительность жизни примерно в 48 лет. Тем не менее, эта цифра вероятно искажена и основана на факте, что этот тип (как все остальные типы синдрома) значительно не выявлен, и высокая пропорция смертельных случаев вызвана посмертным диагностированием. Повышение осведомлённости среди врачей и населения может помочь сделать эту цифру более точной, и сократить число преждевременных смертей. Признаки и симптомы:
- Гиперподвижность, наиболее очевидна на пальцах рук и ног
- Хрупкие стенки сосудов оболочек органов и нежной кожи, имеют склонность к разрыву или образованию аневризмы
- Больные как правило имеют тонкую, бледную и прозрачную кожу (можно видеть вены на груди)
- Артериальная/кишечная/маточная хрупкость или трещины, разрывы
- обширные кровоподтёки
- Некоторые пациенты выражают характерные черты лица (большие глаза, маленький подбородок, тонкий нос и губы, мягкие уши) и имеют маленький рост
В результате возможности маточного разрыва, беременность может оказаться опасной для жизни. Доступно лабораторное тестирование. Кожная биопсия может служить доказательством аномальной структуры коллагена. Этот биомеханический анализ выявляет более 95 % случаев. Лабораторное тестирование рекомендуется лицам имеющим два или более значительных симптома. ДНК анализ может выявить изменения в пределах гена COL3A1. Эта информация может помочь при предродовой генетической консультации, когда один из родителей был диагностирован и известна его генетическая мутация или был продемонстрирован биомеханический дефект.
Тип кифосколиоз
В прежней классификации тип VI. Он очень редок, обнаружено немногим более 60 случаев, передаётся аутосомно-рецессивным механизмом. Основным симптом является общая нестабильность (непрочность) суставов. У младенцев наблюдается слабый мышечный тонус, задержка в развитии моторных навыков, прогрессирующее в течение жизни ненормальное искривление позвоночника сколиоз, при котором обычно больные не могут ходить к 20 годам. Легко ранимые глаза и кожа, также вероятна уязвимость кровеносных сосудов. Наблюдаются: спонтанная отслойка сетчатки, кровоизлияния в стекловидное тело, разрывы глазного яблока и роговицы, склер. Также у костей может быть снижена плотность. Существует четыре основных медицинских критерия при диагностике этого типа. Это гиперподвижные суставы, слабый мышечный тонус у новорождённых, прогрессирующий с рождения сколиоз и хрупкие (уязвимые) глаза, что может придать голубоватый оттенок склерам или вызвать разрыв глаза. Вызван изменениями в хромосоме 1 гена PLOD, который кодирует фермент Лизин Гидролаза[2]. Возможен лабораторный тест, в нём измеряется содержание в моче hydroxylysyl pryridinoline’а. Он крайне чувствителен и специфичен для данного типа синдрома, рекомендуется младенцам с тремя и более основными симптомами. Внутриутробный анализ применяется, если известно о существующем риске: диагностированы больные члены семьи, имеющие положительные результаты тестирования. Вышеупомянутый амниоцентез может быть выполнен, если зародышевые клетки берутся из амниотической жидкости и измерена активность фермента.